- + Merck Millipore
- + Thermo Fisher
- + IKA
- + 显微镜
- + 光度计
- + 成像系统
- + MACS Miltenyi
- + 均质机
- + 粒子计数器
- + 冻干机
- + 灭菌系统
- + 细胞
- + 微量移液工作站
- + 振荡培养箱
- + 生物反应器
- + 切片机
- + 培养箱
- + 蠕动泵
- + 细胞破(粉)碎机
- + 转印膜
- + 超滤管
- + Pellicon 超滤系统
- + 超低温冰箱
- + 清洗机
- + 干燥机
- + 洗瓶机
- + 离心泵
- + 容积泵
- + 各种阀
- + 酶标仪
- + 洗衣板
- + 旋光仪
- + 折光仪
- + 行星球磨机
- + 振动筛
- + 基因导入仪
- + 手套系列
- + 接头\\连接器
- + 培养板/培养瓶
- + 温度控制系统
- + 制冷器
- + 存取系统
- + 轧盖机
- + 细胞因子
- + 细胞分选仪
- + 生物安全柜
- + 渗透压仪
- + 拉曼光谱仪
- + 电泳系统
- + 纯水系统
- + 萃取仪
- + 谱新生物
- + TA 仪器
- + wako
第一期 || 浅谈IVD行业公用系统监测和微生物检测法规
更新时间:2019-12-03 浏览次数:2773
国庆长假已经结束~祝贺大家喜提2019年所有法定节假日,接下来又该投入忙碌的科研工作生活了!
小编在假期前的推文中留了一个“小尾巴”——“基于样品检测量多少的变更需要进行适用性检查吗?”
(点击回顾→ “国庆堵”vs“滤膜堵”)
对于这个问题,我们的建议是:由于原来10ml的供试液较1ml的药浓更高,如含抑菌成份则抑菌性更强,所以在方法适用性检查上能包括浓度更低的供试液,所以建议只需走公司内部规定的变更-修改SOP和记录流程即可,不需要重新进行验证。
解决完“遗留问题”,接下来本文将以法规要求作为指导帮助大家梳理IVD行业中关于微生物监测的方法。
医疗器械生产质量管理规范(GMP)在2015年10月份生效,其中有三大附录:《无菌医疗器械》、《植入性医疗器械》、《体外诊断试剂》。
下面我将整体分析IVD行业关于公用系统和检测样品方面的微生物控制。
➤《医疗器械生产质量管理规范附录体外诊断试剂》
2.2.3应当根据体外诊断试剂的生产过程控制,确定在相应级别的洁净室(区)内进行生产的过程,避免生产中的污染。空气洁净级别不同的洁净室(区)之间的静压差应当大于5帕,洁净室(区)与室外大气的静压差应大于10帕,并应当有指示压差的装置。相同级别洁净室间的压差梯度应当合理。
2.2.5阴性或阳性血清、质粒或血液制品等的处理操作,生产区域应当不低于10000级洁净度级别,并应当与相邻区域保持相对负压。
2.2.11洁净室(区)的温度和相对湿度应当与产品生产工艺要求相适应。无特殊要求时,温度应当控制在18~28℃,相对湿度控制在45%~65%。
2.2.19进行危险度二级及以上的病原体操作应当配备生物安全柜,空气应当进行过滤处理后方可排出。应当对过滤器的性能进行定期检查以保证其有效性。使用病原体类检测试剂的阳性血清应当有相应的防护措施。
2.2.20对于特殊的高致病性病原体的采集、制备,应当按照有关部门颁布的行业标准,如人间传染病微生物名录、微生物和生物医学实验室生物安全通用准则、实验室生物安全通用要求等相关规定,配备相应的生物安全设施。
2.3.1洁净室(区)空气净化系统应当经过确认并保持连续运行,维持相应的洁净度级别,并在一定周期后进行再确认。若停机后再次开启空气净化系统,应当进行必要的测试或验证,以确认仍能达到规定的洁净度级别要求。
2.6.9应当制定洁净室(区)的卫生管理文件,按照规定对洁净室(区)进行清洁处理和消毒,并做好记录。所用的消毒剂或消毒方法不得对设备、容器具、物料和产品造成污染。消毒剂品种应当定期更换,防止产生耐药菌株。
2.7.2生产和检验用的菌毒种应当标明来源,验收、储存、保管、使用、销毁应执行国家有关医学微生物菌种保管的规定和病原微生物实验室生物安全管理条例。应当建立生产用菌毒种的原始种子批、主代种子批和工作种子批系统。
以下为内容大纲,本文将从三个方面进行分析梳理,今天先从“洁净度监测”方面展开。
▶1、洁净度监测
2、温湿度和压差监测
3、病原微生物实验室
一、洁净度监测
▶ 相关法规
《医疗器械生产质量管理规范》
《GB 50591-2010 洁净室施工及验收规范》
《GBT 16292-2010 医药工业洁净室(区)悬浮粒子的测试方法》
《GBT 16293-2010 医药工业洁净室(区)浮游菌的测试方法》
《GBT 16294-2010 医药工业洁净室(区)沉降菌的测试方法》
《ISO 14644-1:2015 洁净室和相关受控环境 第一部分:根据粒子浓度划分空气洁净度等级》
《2010年药品GMP指南:无菌药品》
▶ 粒子监测
洁净区参考《GB 50591-2010》的要求,按照《GBT 16292-2010》方法对粒子进行监测,级别为百级、万级、十万级,生产或检测区域洁净度不低于十万级。
根据《GBT 16292-2010》描述,该法规是参照《ISO 14644-1》,现新版为2015版,建议按《ISO 14644-1:2015》执行,较《ISO 14644-1:1999》和《GBT 16292-2010》,其在取样点数、采样次数、zui小取样量、结果判定、取样管长度、粒子计算器符合要求均有所不同。
▲《GBT 16292-2010》
▶ 沉降菌和浮游菌监测
培养基和时间参考《GBT 16294-2010》,TSA在30-35℃培养不小于2天,SDA在20-25℃培养不小于5天;但zui少采样点数目不应参考《GBT 16294-2010》,应参考《ISO 14644-1:2015》。


▲《GBT 16294-2010》
▶ 标准方面
参照医疗器械GMP,以下是沉降菌动静态监测0.5小时的结果,而粒子数结果判定不需要计算95%UCL;
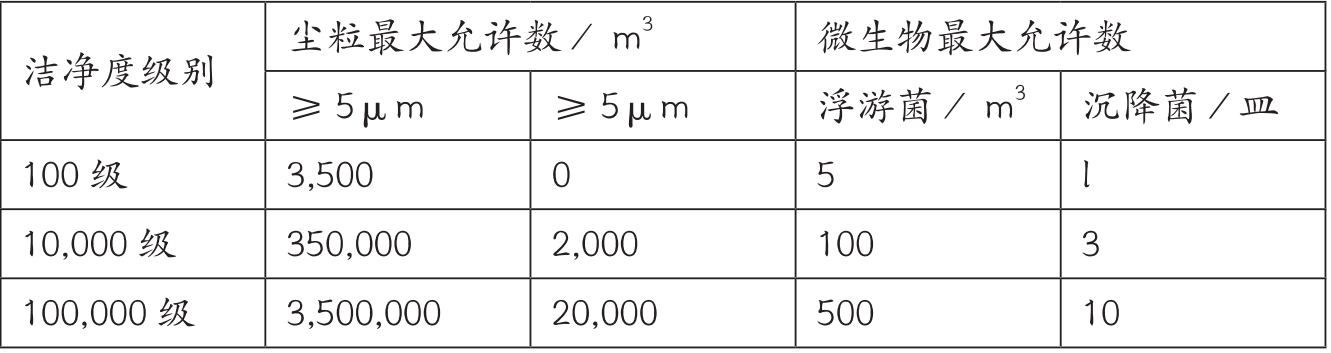
推荐使用Merck环境监测成品培养基和MAS-100 NT浮游菌采样器


▶ 监测频率
《医疗器械生产质量管理规范附录体外诊断试剂》
2.3.1洁净室(区)空气净化系统应当经过确认并保持连续运行,维持相应的洁净度级别,并在一定周期后进行再确认。若停机后再次开启空气净化系统,应当进行必要的测试或验证,以确认仍能达到规定的洁净度级别要求。
静态监测要求参考《GB 50591-2010》,动态监测根据用户风险评估制定;
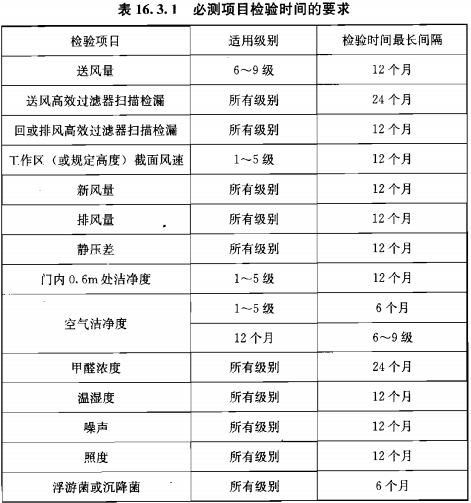
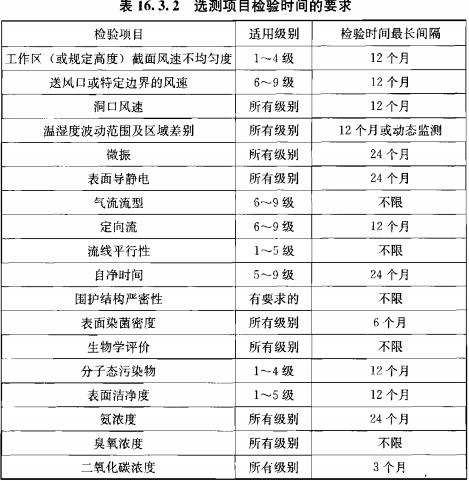
建议动态监测参考2010年药品GMP指南:无菌药品,百级每批生产,万级、十万级每周一次;
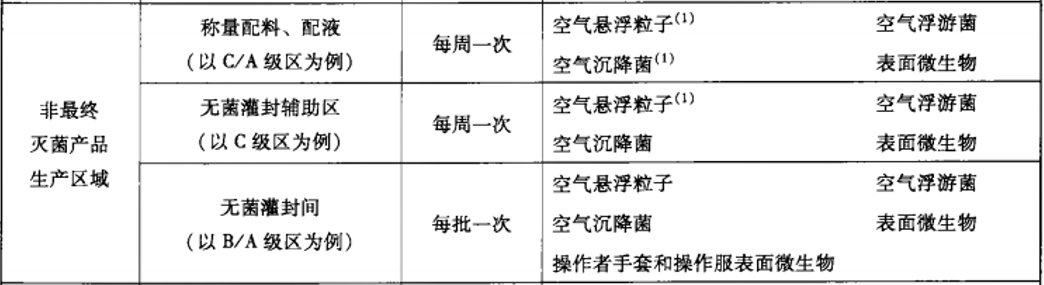
推荐使用TSI 9500系统便携式粒子计数器,有28.3L、50L、100L型号,用于百级、万级、十万级洁净区的尘埃粒子监测。内含多种标准《ISO14644-1:1999》、《EUGMP-ISO:1999》、《ISO14644-1:2015》、《EUGMP-ISO:2015》,适合制药行业和医疗器械行业的使用。输入”面积”、“洁净等级”、“选择标准”和“房间状态”后自动生成“通道”、“取样量”、“位点数”、“取样时间”等的数据,其数据完整性符合FDA 21 CFR PART11、Part211和212要求;

以上就是“洁净度监测”的分析内容,下期将围绕“温湿度和压差监测”展开分析梳理,敬请期待!
- (上一篇):想除热原?买它!!
- (下一篇):生物安全柜怎么用更安全?